Hypertension artérielle pulmonaire
L'hypertension pulmonaire ou PH est une affection caractérisée par une pression artérielle élevée dans les poumons. Cette condition rend la respiration difficile. Certaines personnes atteintes ont besoin d'un supplément d'oxygène. Cette affectio…
L'hypertension pulmonaire ou PH est une affection caractérisée par une pression artérielle élevée dans les poumons. Cette condition rend la respiration difficile. Certaines personnes atteintes ont besoin d'un supplément d'oxygène. Cette affection peut également provoquer des vertiges et une fatigue importante. Certaines personnes atteintes s'évanouissent facilement. Les symptômes s'aggravent lorsqu'elles font de l'exercice ou travaillent dur. L'hypertension pulmonaire est une maladie grave, qui peut être mortelle. Elle rend le cœur plus difficile à pomper le sang. Comme le cœur doit travailler plus dur, il peut aussi tomber malade. Certaines personnes très malades peuvent avoir besoin d'une transplantation pulmonaire ou d'une transplantation cœur-poumon pour vivre. Hypertension pulmonaire : son nom complet est hypertension artérielle pulmonaire, même si la plupart des gens l'appellent pah, ph ou pha.
Galerie d’images
9 Images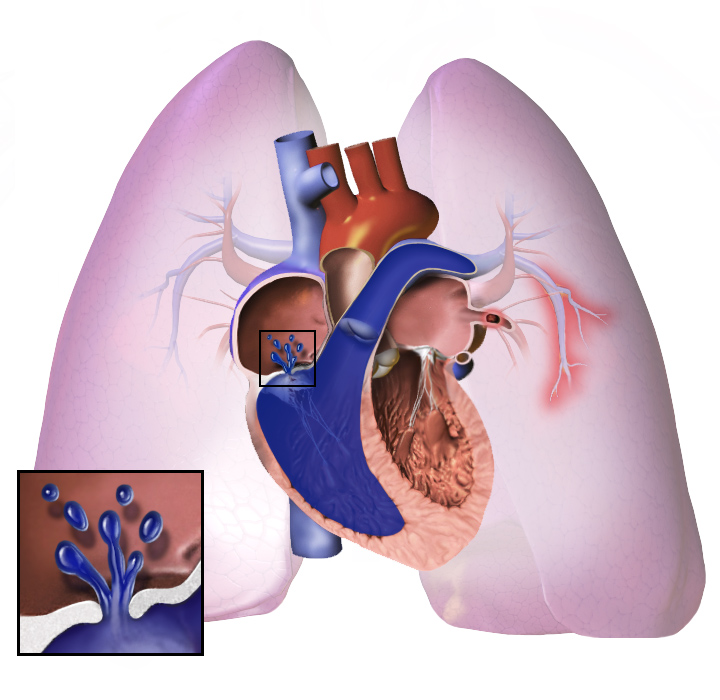






Signes et symptômes
Les personnes souffrant d'hypertension pulmonaire ont des difficultés à respirer. Elles se fatiguent aussi facilement. Certaines d'entre elles s'évanouissent aussi facilement. Elles peuvent avoir des douleurs thoraciques. Certains patients présentent un gonflement des pieds et des chevilles. Ces symptômes s'aggravent pendant l'exercice ou le travail.
Comme de nombreuses maladies peuvent rendre la respiration difficile, le médecin doit connaître les antécédents du patient. Cela l'aide à traiter le patient, même si celui-ci souffre d'une autre maladie. Le médecin procède également à plusieurs examens. L'hypertension pulmonaire fait que le cœur sonne différemment. L'un des tests consiste à mesurer la pression sanguine à l'intérieur de l'artère pulmonaire, le vaisseau sanguin qui va du cœur aux poumons.
Afin d'établir la cause, le médecin procède généralement à un examen approfondi des antécédents médicaux. Une histoire familiale détaillée est prise pour déterminer si la maladie peut être familiale. Des antécédents d'exposition à la cocaïne, à la méthamphétamine, à l'alcool entraînant une cirrhose et au tabac entraînant un emphysème sont considérés comme importants. Un examen physique est effectué pour rechercher les signes typiques de l'hypertension pulmonaire, notamment un fort P2 (bruit de fermeture de la valve pulmonaire), un soulèvement (para)sternal, une distension de la veine jugulaire, un œdème de la pédale, une ascite, un reflux hépatojugulaire, des coups de matraque, etc.
Ce qui ne va pas avec le corps
En cas d'hypertension pulmonaire, les vaisseaux sanguins des poumons deviennent trop étroits. La pression sanguine dans les poumons devient élevée. Le cœur travaille très dur pour pomper le sang dans les vaisseaux sanguins étroits. Plus tard, les vaisseaux sanguins des poumons deviennent durs et épais. Le cœur doit travailler plus fort.
Le cœur peut travailler si dur qu'il en devient malade. C'est ce qu'on appelle l'insuffisance cardiaque. Le cœur malade ne peut pas bien pomper le sang. Moins de sang va aux poumons, donc le sang reçoit moins d'oxygène. Il a alors du mal à respirer. La situation s'aggrave lorsqu'on fait de l'exercice ou qu'on travaille dur.
Causes
La cause la plus fréquente de l'hypertension pulmonaire est l'insuffisance cardiaque gauche. Celle-ci provoque une hypertension artérielle veineuse pulmonaire. Cela entraîne un œdème pulmonaire, ou une accumulation de liquide dans les poumons.
De nombreuses maladies peuvent provoquer une hypertension artérielle pulmonaire (HAP).
- Les maladies pulmonaires qui font que le sang contient moins d'oxygène, comme :
· la maladie pulmonaire obstructive chronique ou BPCO
· maladie pulmonaire interstitielle
· Le syndrome de Pickwicki
- les problèmes du système immunitaire, tels que :
· SIDA
· sclérodermie
· autres troubles auto-immuns
- les problèmes de foie
· cirrhose
· hypertension portale
- autres causes
· l'apnée du sommeil
· prendre des pilules pour perdre du poids, comme le Fen-Phen, l'Aminorex, la fenfluramine (Pondimin) et la phentermine
· la drépanocytose,
· maladie cardiaque congénitale
· les maladies de la thyroïde,
· la prise de drogues comme la cocaïne
· peut-être l'herpèsvirus humain 8
Lorsqu'une personne souffre d'hypertension pulmonaire sans autre cause, on parle d'hypertension artérielle pulmonaire idiopathique ou HAPI.
Lorsqu'il existe des antécédents familiaux, la maladie est appelée hypertension artérielle pulmonaire familiale (HAPF). L'HAPI et l'HAPP sont désormais considérées comme des maladies génétiques liées à des mutations du gène BMPR2, qui code pour un récepteur des protéines morphogénétiques osseuses, ainsi que du gène 5-HT(2B), qui code pour un récepteur de la sérotonine.
En médecine, l'hypertension pulmonaire (HP) est une augmentation de la pression sanguine dans l'artère pulmonaire ou le système vasculaire des poumons, entraînant un essoufflement, des étourdissements, des évanouissements et d'autres symptômes, tous exacerbés par l'effort. Selon la cause, l'hypertension pulmonaire peut être une maladie grave avec une tolérance à l'effort nettement réduite et une insuffisance cardiaque du côté droit. Elle a été identifiée pour la première fois par le Dr Ernst von Romberg en 1891. Elle peut être de cinq types différents : artérielle, veineuse, hypoxique, thromboembolique ou diverse.
Bien que les termes "hypertension pulmonaire primaire" (qui signifie de cause inconnue) et "hypertension pulmonaire secondaire" (qui signifie due à une autre maladie) persistent encore dans les documents diffusés aux patients et au grand public, ces termes ont été largement abandonnés dans la littérature médicale. Ce changement s'est produit parce que l'ancienne classification dichotomique ne reflétait pas la pathophysiologie ou le résultat. Elle a conduit à des décisions thérapeutiques erronées, c'est-à-dire à ne traiter que l'hypertension pulmonaire "primaire". Cela a conduit au nihilisme thérapeutique pour de nombreux patients qualifiés d'hypertension pulmonaire "secondaire", et pourrait avoir contribué à leur décès. L'expression "hypertension pulmonaire primaire" a été remplacée par "hypertension artérielle pulmonaire idiopathique". Les termes "hypertension pulmonaire primaire" et "secondaire" ne doivent plus être utilisés. Vous trouverez de plus amples informations dans la section "Classification" ci-dessous.
Causes
La cause la plus fréquente de l'hypertension pulmonaire est l'insuffisance cardiaque gauche, qui entraîne une hypertension veineuse pulmonaire. Celle-ci peut être due à un dysfonctionnement systolique ou diastolique du ventricule gauche ou à un dysfonctionnement valvulaire tel qu'une régurgitation mitrale ou une sténose mitrale. Elle se manifeste généralement par un œdème pulmonaire.
Les causes courantes de l'hypertension artérielle pulmonaire (HAP) sont notamment le VIH, la sclérodermie et d'autres maladies auto-immunes, la cirrhose et l'hypertension portale, la drépanocytose, les cardiopathies congénitales et les maladies thyroïdiennes. L'utilisation de pilules amaigrissantes telles que le Fen-Phen, l'Aminorex, la fenfluramine (Pondimin) et la phentermine a conduit au développement de l'HAP dans le passé.
Pathogenèse
Quelle que soit la cause initiale, l'hypertension pulmonaire implique le resserrement des vaisseaux sanguins reliés aux poumons et à l'intérieur de ceux-ci. Il est donc plus difficile pour le cœur de pomper le sang dans les poumons, tout comme il est plus difficile de faire circuler l'eau dans un tuyau étroit que dans un tuyau large. Avec le temps, les vaisseaux sanguins affectés deviennent à la fois plus rigides et plus épais, ce qui augmente encore la pression sanguine dans les poumons et entrave la circulation sanguine. En outre, la charge de travail accrue du cœur entraîne un épaississement et un élargissement du ventricule droit, ce qui rend le cœur moins apte à pomper le sang dans les poumons, ce qui provoque une insuffisance cardiaque droite. À mesure que le flux sanguin dans les poumons diminue, le côté gauche du cœur reçoit moins de sang. Ce sang peut également transporter moins d'oxygène que la normale. Par conséquent, il devient de plus en plus difficile pour le côté gauche du cœur de pomper pour fournir suffisamment d'oxygène au reste du corps, en particulier pendant l'activité physique.
Diagnostic
L'hypertension pulmonaire pouvant être de 5 types principaux, une série de tests doit être effectuée pour distinguer l'hypertension artérielle pulmonaire des variétés veineuses, hypoxiques, thomboemboliques ou diverses.
Un examen physique est effectué pour rechercher les signes typiques de l'hypertension pulmonaire. Ces signes comprennent des bruits cardiaques altérés, tels qu'un son S2 ou un deuxième son cardiaque largement divisé, un P2 fort ou un son de fermeture de la valve pulmonaire (faisant partie du deuxième son cardiaque), un soulèvement (para)sternal, un possible son S3 ou un troisième son cardiaque, et une régurgitation pulmonaire. Les autres signes comprennent la distension de la veine jugulaire (élargissement des veines jugulaires), l'œdème périphérique (gonflement des chevilles et des pieds), l'ascite (gonflement abdominal dû à l'accumulation de liquide), le reflux hépatojugulaire et le matraquage.
D'autres procédures sont nécessaires pour confirmer la présence d'une hypertension pulmonaire et exclure d'autres diagnostics possibles. Il s'agit généralement de tests de la fonction pulmonaire, d'analyses sanguines, d'électrocardiographie (ECG), de mesures des gaz du sang artériel, de radiographies de la poitrine (suivies d'un scanner à haute résolution si l'on soupçonne une maladie pulmonaire interstitielle), et d'une ventilation-perfusion ou d'un scanner V/Q pour exclure une hypertension pulmonaire thromboembolique chronique. Une biopsie du poumon n'est généralement pas indiquée, sauf si l'on pense que l'hypertension pulmonaire est due à une maladie pulmonaire interstitielle sous-jacente. Mais les biopsies pulmonaires comportent des risques d'hémorragie en raison de la pression artérielle intrapulmonaire élevée. L'amélioration clinique est souvent mesurée par un "test de marche de six minutes", c'est-à-dire la distance qu'un patient peut parcourir à pied en six minutes. La stabilité et l'amélioration de cette mesure sont en corrélation avec une meilleure survie.
Bien que la pression artérielle pulmonaire puisse être estimée sur la base d'une échocardiographie, le prélèvement de pression avec un cathéter de Swan-Ganz fournit la mesure la plus précise. La PAOP et la PVR ne peuvent pas être mesurées directement avec l'échocardiographie. Par conséquent, le diagnostic de l'HAP nécessite un cathétérisme cardiaque. Un cathéter de Swan-Ganz peut également mesurer le débit cardiaque, qui est bien plus important pour mesurer la gravité de la maladie que la pression artérielle pulmonaire.
La pression artérielle pulmonaire normale chez une personne vivant au niveau de la mer a une valeur moyenne de 12-16 mm Hg (1600-2100 Pa). Une hypertension pulmonaire est définie lorsque la pression moyenne au repos dépasse 25 mm Hg (3300 Pa). Si la pression artérielle pulmonaire moyenne dépasse 30 mm Hg (4000 Pa) à l'effort, on parle également d'hypertension pulmonaire.
Le diagnostic d'HAP nécessite la présence d'une hypertension pulmonaire avec deux autres affections. La pression d'occlusion des artères pulmonaires (PAOP ou PCWP) doit être inférieure à 15 mm Hg (2000 Pa) et la résistance vasculaire pulmonaire (PVR) doit être supérieure à 3 unités Wood (240 dyn-s-cm-5 ou 2,4 mN-s-cm-5).
Classification
Classification actuelle
En 2003, le 3e symposium mondial sur l'hypertension artérielle pulmonaire a été organisé à Venise pour modifier la classification en fonction de la nouvelle compréhension des mécanismes de la maladie. Le système révisé élaboré par ce groupe fournit le cadre actuel pour comprendre l'hypertension artérielle pulmonaire.
Le système comporte plusieurs améliorations par rapport à l'ancien système de classification d'Évian de 1998. Les descriptions des facteurs de risque ont été mises à jour et la classification des shunts congénitaux systémiques à pulmonaires a été révisée. Une nouvelle classification des facteurs génétiques dans le PH a été recommandée, mais n'a pas été mise en œuvre car les données disponibles ont été jugées inadéquates.
Le système de classification révisé de Venise 2003 peut être résumé comme suit :
- Groupe I de l'OMS - Hypertension artérielle pulmonaire (HAP)
- Groupe II de l'OMS - Hypertension pulmonaire associée à une maladie cardiaque gauche
- Groupe III de l'OMS - Hypertension pulmonaire associée à des maladies pulmonaires et/ou à l'hypoxémie
- Groupe IV de l'OMS - Hypertension pulmonaire due à une maladie thrombotique et/ou embolique chronique
- Groupe V de l'OMS - Divers
Terminologie précédente
Les termes d'hypertension pulmonaire primaire et secondaire (HPP et HSP) étaient autrefois utilisés pour classer la maladie. Cela a conduit à l'hypothèse que seule la maladie primaire devait être traitée, et que la variété secondaire devait être ignorée au profit du traitement de la maladie sous-jacente. En fait, toutes les formes d'hypertension artérielle pulmonaire sont traitables. Malheureusement, ce système de classification persiste encore dans l'esprit de nombreux médecins, et conduit probablement à refuser le traitement à de nombreux patients. Cette approche nihiliste de l'hypertension artérielle pulmonaire peut également contribuer à un sous-diagnostic. On estime qu'il y a environ 100 000 patients atteints d'HAP aux États-Unis, mais seuls 15 à 20 000 d'entre eux ont été diagnostiqués. Beaucoup d'autres ont été mal diagnostiqués comme souffrant de BPCO, d'asthme ou d'insuffisance cardiaque congestive.
Le terme d'hypertension pulmonaire primaire (HPP) a maintenant été remplacé par celui d'hypertension artérielle pulmonaire idiopathique (HAPI) dans une grande partie de la littérature médicale. Cependant, certains médecins continuent d'utiliser l'ancienne classification de manière inappropriée.
Epidémiologie
L'IPAH est une maladie rare dont l'incidence est d'environ 2 à 3 par million par an et la prévalence d'environ 15 par million. Les femmes sont presque trois fois plus susceptibles de présenter une HAP que les hommes.
Les autres formes d'HAP sont beaucoup plus courantes. Dans la sclérodermie, l'incidence a été estimée entre 6 et 60 % de tous les patients, dans la polyarthrite rhumatoïde jusqu'à 21 %, dans le lupus érythémateux systémique entre 4 et 14 %, dans l'hypertension portale entre 2 et 5 %, dans le VIH environ 0,5 % et dans la drépanocytose entre 20 et 40 %.
Les pilules diététiques comme le Fen-Phen ont produit une incidence annuelle de 25 à 50 par million par an.
Traitement
Le traitement est déterminé par le fait que le PH est artériel, veineux, hypoxique, thromboembolique ou divers. L'hypertension veineuse pulmonaire étant synonyme d'insuffisance cardiaque congestive, le traitement consiste à optimiser la fonction ventriculaire gauche par l'utilisation de diurétiques, de bêta-bloquants, d'inhibiteurs de l'ECA, etc. ou à réparer/remplacer la valve mitrale ou aortique.
Dans l'HAP, les changements de mode de vie, la digoxine, les diurétiques, les anticoagulants oraux et l'oxygénothérapie sont considérés comme des traitements classiques, mais leur effet bénéfique n'a jamais été prouvé de manière aléatoire et prospective.
Les inhibiteurs calciques à forte dose ne sont utiles que chez 5 % des patients atteints d'IPAH qui sont vasoréactifs au cathéter de Swan-Ganz. Malheureusement, les inhibiteurs calciques ont été largement mal utilisés, étant prescrits à de nombreux patients atteints d'HAP non vasoréactive, ce qui entraîne une morbidité et une mortalité excessives.
Substances vasoactives
Trois voies principales sont impliquées dans la prolifération et la contraction anormales des cellules musculaires lisses de l'artère pulmonaire chez les patients souffrant d'hypertension artérielle pulmonaire. Ces voies correspondent à des cibles thérapeutiques importantes dans cette maladie et jouent un rôle dans la détermination des trois classes de médicaments qui seront utilisés : les antagonistes des récepteurs de l'endothéline, les inhibiteurs de la phosphodiestérase de type 5 et les dérivés de la prostacycline.
La prostacycline (prostaglandine I2) est généralement considérée comme le traitement le plus efficace contre l'HAP. L'époprosténol (prostacycline synthétique, commercialisée sous le nom de Flolan®) est administré par perfusion continue qui nécessite un cathéter veineux central semi-permanent. Ce système d'administration peut provoquer un sepsis et une thrombose. Le Flolan® est instable et doit donc être conservé sur de la glace pendant l'administration. Comme il a une demi-vie de 3 à 5 minutes, la perfusion doit être continue (24 heures sur 24, 7 jours sur 7), et l'interruption peut être fatale. D'autres prostanoïdes ont donc été mis au point. Le tréprostinil (Remodulin®) peut être administré par voie intraveineuse ou sous-cutanée, mais la forme sous-cutanée peut être très douloureuse. L'ilomédine (Iloprost®) est également utilisée en Europe par voie intraveineuse et a une demi-vie plus longue. L'Iloprost (commercialisé sous le nom de Ventavis®) est la seule forme inhalée de prostacycline dont l'utilisation est autorisée aux États-Unis et en Europe. Cette forme d'administration présente l'avantage d'un dépôt sélectif dans les poumons avec moins d'effets secondaires systémiques.
Le bosentan, antagoniste double (ETA et ETB) des récepteurs de l'endothéline (commercialisé sous le nom de Tracleer®), a été approuvé en 2001. Deux antagonistes sélectifs des récepteurs de l'endothéline (ETA uniquement) sont en phase finale d'approbation : le sitaxsentan et l'ambrisentan. Le sildénafil, un inhibiteur sélectif de la phosphodiestérase de type 5 (PDE5) spécifique aux BPF, a été approuvé pour le traitement des HAP en 2005. Il est commercialisé pour les HAP sous le nom de Revatio®. Le tadalafil (actuellement commercialisé sous le nom de Cialis® pour le traitement des troubles de l'érection) est actuellement en phase III des essais. Le peptide intestinal vasoactif par inhalation devrait entrer en essais cliniques pour l'HAP en 2007. Le PRX-08066 est un antagoniste de la sérotonine actuellement en cours de développement pour l'hypertension pulmonaire hypoxique.
Chirurgical
La septostomie auriculaire est une procédure chirurgicale qui crée une communication entre les oreillettes droite et gauche. Elle soulage la pression du côté droit du cœur, mais au prix d'une baisse du taux d'oxygène dans le sang (hypoxie). Il est préférable de la pratiquer dans des centres expérimentés. La transplantation pulmonaire guérit l'hypertension artérielle pulmonaire, mais laisse le patient avec les complications de la transplantation et une survie d'environ 5 ans.
La thromboendarterectomie pulmonaire (PTE) est une procédure chirurgicale utilisée pour l'hypertension pulmonaire thromboembolique chronique. Il s'agit de l'ablation chirurgicale d'un thrombus organisé (caillot) avec la paroi de l'artère pulmonaire ; c'est une procédure importante et très difficile qui est actuellement pratiquée dans quelques centres sélectionnés. Des séries de cas montrent un succès remarquable chez la plupart des patients.
Le traitement des variétés hypoxiques et diverses de l'hypertension pulmonaire n'a pas été établi. Toutefois, des études sur plusieurs agents sont en cours pour recruter des patients. De nombreux médecins traiteront ces maladies avec les mêmes médicaments que pour l'HTAP, jusqu'à ce que de meilleures options soient disponibles.
Pronostic
Le registre IPAH du NIH des années 1980 a montré une survie médiane non traitée de 2 à 3 ans à partir du diagnostic, la cause du décès étant généralement une insuffisance ventriculaire droite (cor pulmonaire). Bien que ce chiffre soit largement cité, il n'est probablement plus pertinent aujourd'hui. Les résultats ont considérablement changé au cours des deux dernières décennies. Cela peut être dû à l'apparition de nouveaux médicaments, à l'amélioration des soins généraux et à un diagnostic plus précoce (biais de délai). Une étude récente sur les résultats des patients qui avaient commencé un traitement au bosentan (Tracleer®) a montré que 86 % des patients étaient en vie à 3 ans. Avec la disponibilité de plusieurs agents, la thérapie combinée est de plus en plus utilisée. L'impact de ces agents sur la survie n'est pas connu, car beaucoup d'entre eux n'ont été développés que récemment. Il ne serait pas déraisonnable de s'attendre à ce que la survie médiane dépasse 10 ans dans un avenir proche.
Questions et réponses
Q : Qu'est-ce que l'hypertension pulmonaire ?
R : L'hypertension pulmonaire est une affection caractérisée par une pression sanguine élevée dans les poumons.
Q : Quels sont les symptômes de l'hypertension pulmonaire ?
R : Les symptômes de l'hypertension pulmonaire comprennent des difficultés respiratoires, des vertiges, de la fatigue et des évanouissements.
Q : Pourquoi certaines personnes souffrant d'hypertension pulmonaire ont-elles besoin de plus d'oxygène ?
R : Certaines personnes atteintes d'hypertension pulmonaire ont besoin d'oxygène supplémentaire parce que leur maladie les empêche de respirer.
Q : Quand les symptômes de l'hypertension pulmonaire s'aggravent-ils ?
R : Les symptômes de l'hypertension pulmonaire s'aggravent lors d'un exercice ou d'un travail intense.
Q : Pourquoi l'hypertension pulmonaire est-elle une maladie grave ?
R : L'hypertension pulmonaire est une affection grave parce qu'elle rend plus difficile le pompage du sang par le cœur et peut être fatale.
Q : Quel est le nom complet de l'hypertension pulmonaire ?
R : Le nom complet de l'hypertension pulmonaire est hypertension artérielle pulmonaire, même si la plupart des gens l'appellent pah, ph ou pha.
Q : De quoi certaines personnes très malades atteintes d'hypertension pulmonaire peuvent-elles avoir besoin pour vivre ?
R : Certaines personnes très malades atteintes d'hypertension pulmonaire peuvent avoir besoin d'une transplantation pulmonaire ou d'une transplantation cœur-poumon pour vivre.
Articles liés
Auteur
AlegsaOnline.com Hypertension artérielle pulmonaire Leandro Alegsa
URL: https://fr.alegsaonline.com/art/80022
Sources
- ncbi.nlm.nih.gov : PMID 8692238
- ncbi.nlm.nih.gov : PMID 14985486
- ncbi.nlm.nih.gov : PMID 10555089
- ncbi.nlm.nih.gov : PMID 13679525
- ncbi.nlm.nih.gov : PMID 10903931
- ncbi.nlm.nih.gov : PMID 14659797
- ncbi.nlm.nih.gov : PMID 15194171